Small molecule force field parametrization for atomistic Molecular Dynamics simulations¶
Overview¶
This use case aims to illustrate the process of parameterizing a small molecule, to be used in a Molecular Dynamics simulation, step by step. The particular example used is the Imipramine molecule (PDB code IXX, DrugBank code DB00458).
Imipramine is a tricyclic antidepressant (TCA) which is used mainly in the treatment of depression. It can also reduce symptoms of agitation and anxiety.
Background¶
Molecular Dynamics (MD) simulation is the most popular theoretical technique to obtain macromolecular dynamic information. Classical mechanics is used to represent atoms as spheres of a given radius, hardness, charge and mass. The energy functional used by force-fields is usually composed of two terms: bonded and non-bonded components:
where
and
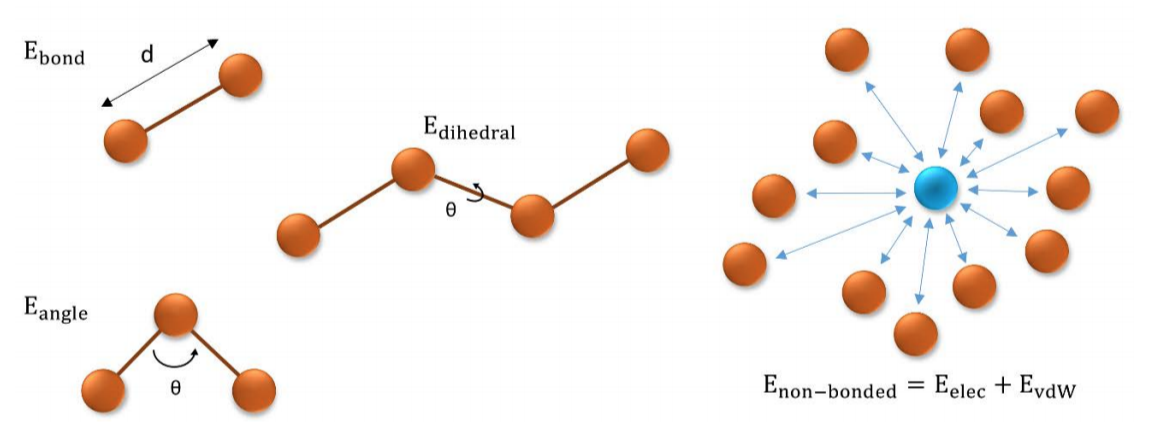
The combination of the force-fields with the laws of classical mechanics (Newton’s second law of motion), allows the calculation of the time evolution of the system. Trajectories of atoms and molecules are determined by numerically solving Newton’s equations of motion for a system of interacting particles, where forces between the particles and their potential energies are calculated using the force-field energy functionals.
Force-field parameters for standard amino-acids and nucleic acids exist and are typically included in MD packages. Unfortunately, this is not the case for small molecules. That makes a ligand parameterization process mandatory if we are interested in simulating a protein-ligand complex.
How it works ?¶
This workflow makes extensive use of the BioExcel Building Blocks library (biobb). Each step of the process is performed by a building block (bb), which are wrappers of tools/scripts that computes a particular functionality (e.g. Solvating a system). If you are interested in expanding/modifying the current workflow, please visit the existing documentation for each of the packages here.
Most of the steps performed in this pipeline run GROMACS MD package tools, one of the most popular MD packages available.
Although the pipeline is presented step by step with associated information, it is extremely advisable to previously spend some time reading documentation about small molecule parameterization, to get familiar with the terms used, especially for newcomers to the field.
Outcomes / Steps¶
This use case will explain:
How to fetch a ligand structure in PDB format from the MMB PDB mirror REST API
How to add hydrogen atoms to the small molecule
How to energetically minimize the hydrogen atoms of the molecule
How to generate the ligand parameters
How to find and download the generated output files
Input¶
A ligand code (3-letter code)
The ligand net charge
The pH, acidity or alkalinity for the small molecule. Hydrogen atoms will be added according to this pH.
Outputs¶
Interactive and 3D vizualisation of the intermediate results on the small molecule
Interactive and 3D vizualisation of the resulting parameterized structure
Collection of parameter files needed to run an MD simulation including the small molecule available to download
Targeted audience¶
All scientists working in biology related areas where protein study is relevant with a focus on structural biologists and biochemists. Especially directed to scientists interested in protein dynamics and flexibility.