Identify potential protein binding sites by comparing the electrostatic potentials of a set of protein isoforms¶
Overview¶
This use case describes how to compare the electrostatic potentials of nine isoforms of the enzyme adenylyl cyclase protein at many sites on its surface. The electrostatic similarities at these sites is compared to find isoform-specific regulation patterns for the inhibitory protein, to predict the likely binding site of regulatory proteins. It uses the multipipsa software tool, which also helps to automate these calculations by providing a python wrapper for the following open source software tools:
PDB2PQR: A tool that takes a protein structure in PDB format, adds missing hydrogen atoms, and creates a structure file in PQR format. The PQR file format is derived from the PDB format for describing atomic data, but with the occupancy and temperature factor fields replaced with atomic partial charges and radii.
APBS: A tool that calculates electrostatic potentials through solution of the Poisson-Boltzmann equation, one of the most common continuum models for describing electrostatic interactions between molecular solutes in salty, aqueous media.
Background¶
For details on computation of the electrostatic potential see Calculate the electrostatic potential of a protein from its atomic structure.
For details on the similarity computations and pipsa algorithm, see Compare the electrostatic potentials surrounding a set of protein isoforms with multipipsa
Input Data¶
In this use case, we use as our input structure a structure of the catalytic domain of the enzyme adenylyl cyclase 5 (AC5), modelled during the work described in Tong et al (2016). The structures of the AC isoforms were created via homology modelling using the same template. The region where there are significant structural differences between the isoforms is in a flexible loop region that was not defined in the template structure. There are also variations in sequence length across AC isoforms in this region.
Procedure¶
Structure of AC5 is visualized. The catalytic domain of AC5 is a dimer consisting of two protein chains. In the full structure of AC5 these two chains are connected by a series of transmembrane helices that anchor the protein in the post-synaptic membrane.
Then the Pdb2Pqr method is used to generate hydrogen atoms in the protein structure. Proteins contain a number of ionisable amino acids, which can exist in different protonation states, depending on the pH of the solution they are in. PDB2PQR can predict the states of these amino acids, at a given pH (defined as 7.4 in the last cell, a normal physiological pH), then add all missing hydrogen atoms to the structure, and assign atomic charges and radii to all atoms. By default, multipipsa assigns charges and radii from the Amber force field.
The APBS method used to solve the linearised Poisson-Boltzmann equation to obtain the electrostatic potential in the dx and UHBD file formats. It also creates a dx file describing the solvent excluded volume of AC5. This is used for visualisation later.
Finally, electrostatic similarity between AC isoforms is computed.
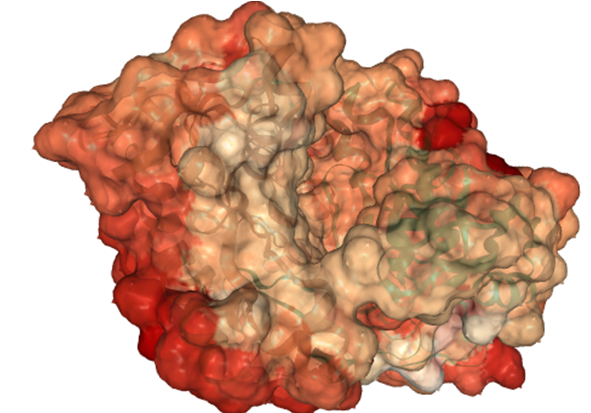
Results¶
The similarity of AC5 isoforms is indicated by the surface color from the most dissimilar regions (\(SI_{12}=-1\), shown in red) to the highly similar regions (\(SI_{12}=1\), shown in white):